What Is Cystic Fibrosis and Who Is Affected?
In the realm of genetic conditions, cystic fibrosis (CF) is a lifelong affliction with devastating effects on the body's systems. The disease causes the body to make thick and sticky mucus that is difficult to clear from the lungs, pancreas and other organs, according to Traci M. Kazmerski, M.D., an assistant professor of pediatrics at the University of Pittsburgh.
Cystic fibrosis affects people around the world and its earliest reports date back thousands of years, though it wasn't a recognized medical condition until 1938.
"The prevalence of cystic fibrosis doesn't change by demographic," said Jyoti Matta, M.D., a pulmonologist affiliated with RWJBarnabas Health Medical Group in Jersey City, New Jersey. "Anyone from any community can have cystic fibrosis based on their parents' genetics. CF is traditionally diagnosed early on."
Unfortunately, the condition leads to progressive deterioration and decreased life expectancy, according to Jo Armstrong, CEO of Cystic Fibrosis Australia. In addition, patients are more likely to have or develop comorbidities.
Causes and effects of cystic fibrosis
The root cause of cystic fibrosis is genetics. It can come from one or both parents if they are carriers of a mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, Matta explained.
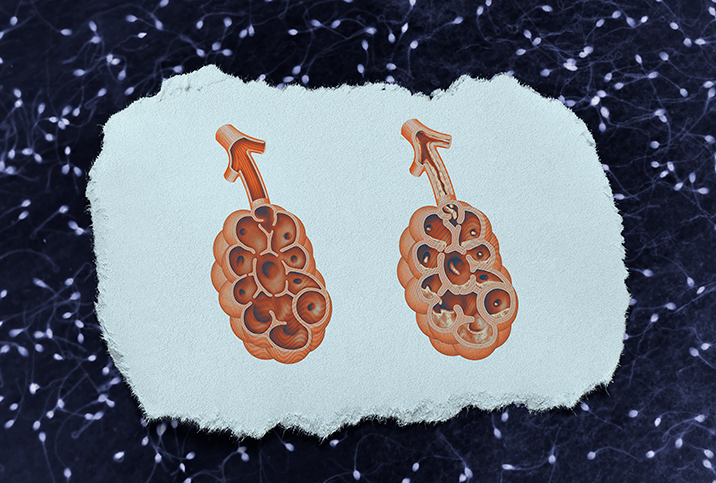
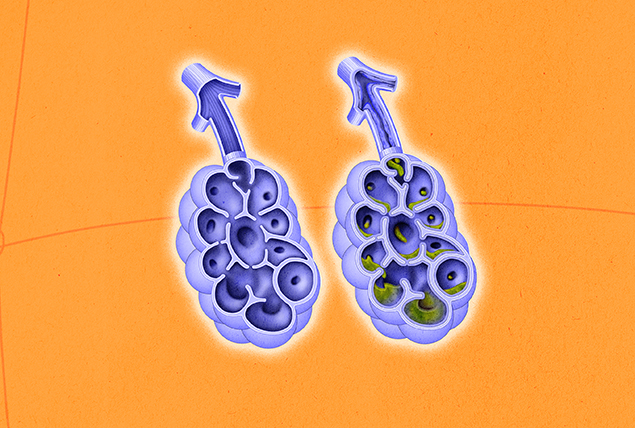
When this gene isn't working properly, it cannot regulate chloride (salt) levels at the cell surface. As a result, water is not attracted to the membrane, leading to the buildup of thick, viscous mucus.
Mutations of these CFTR proteins can affect multiple areas because they lie within the exocrine system, which is the epicenter of all mucus-producing organs. The most prominent effects are on the respiratory and digestive systems, and it is not uncommon for people with CF to require organ transplants.
The overproduction of mucus acts similarly to a drain clog, preventing substances from moving efficiently throughout the body and trapping additional germs inside it. This puts people with cystic fibrosis at an elevated risk for infections.
Airway flow is significantly compromised, and proteins and other nutrients cannot reach the proper areas for absorption, which leads to nutritional deficits and stunted growth.
Over time, cystic fibrosis symptoms get worse and are eventually fatal. Death is usually caused by respiratory complications. More severe cases tend to be apparent at birth, but milder cases may not present any symptoms until the sufferer reaches adulthood.
Types and stages of cystic fibrosis
Cystic fibrosis has diversified levels of intensity.
"The disease can be mild, moderate or severe, depending on the degree to which the cystic fibrosis transmembrane conductance regulator gene is mutated," Matta said.
Researchers have identified more than 1,700 mutations that cause CF, all with assorted strengths. They have further categorized them into six classes:
- Class I. No functional CFTR proteins are made.
- Class II. CFTR proteins are made but cannot reach the cell surface.
- Class III. CFTR proteins reach the cell surface but the channel gate doesn't open.
- Class IV. CFTR proteins make it to the cell surface but the channel gate is dysfunctional.
- Class V. CFTR proteins travel to the cell surface but in insufficient quantities.
- Class VI. CFTR proteins are made but work incorrectly at the cell membrane.
Class I is the most critical type because virtually no CFTR is present, and the degree of severity descends from there. Most people with CF fall into the first three categories and have a worse prognosis than those who fall within the second three. They are typically younger than those in classes IV through VI and have elevated incidences of liver disease, CF-related diabetes and acid reflux.
Patients in classes III to VI tend to experience milder lung and pancreatic complications but are more likely to have osteoporosis and pancreatitis. They have longer average life spans than their counterparts. These cases often achieve symptom management with medications that modify the function of the gene modulator, Matta said.
Socioeconomic and cultural influences
White people are most prone to a cystic fibrosis diagnosis. However, the condition is on the rise for minorities, and research suggests they have more negative health outcomes. Hispanics, in particular, are at risk because they have CFTR mutations that are anomalous from those seen in non-Hispanics and are, thus, insufficiently affected by targeted treatments. Consequently, they have higher hospitalization and mortality rates.
Black CF patients don't have the same unique mutations but still experience similar upward trends of hospitalizations and death.
Socioeconomic status (SES) is impactful on CF outcomes, too. High SES is associated with higher rates of diagnoses but better prognoses because of treatment availability. People of low SES have less access to adequate healthcare and lack representation in cystic fibrosis registries. They are sometimes unable to afford costly medications and therapies due to insurance barriers.
Regardless of the disease's severity, patients with more socioeconomic barriers have decreased chances of being placed on an organ transplant waiting list. Low SES also has been correlated with poorer medication adherence and respiratory functioning in children.
Environmental conditions are important because exposure to certain chemicals and tobacco smoke leads to worse nutritional and pulmonary results.
Facts, statistics and studies
Cystic fibrosis is seen worldwide but is most prevalent in European countries, followed closely by the United States. Both have incidence rates of 1 in 2,000 to 3,000 live births. Australia has comparable rates at 1 in 2,500 live births, and about 1 in 25 people are genetic carriers, according to Armstrong.
It is thought that cystic fibrosis goes underdiagnosed in Africa and Asia due to a lack of testing and documentation, along with potentially inaccurate diagnostic rates, which is why its prevalence seems so low.
Modern medicine has yielded an upward trend in life expectancy for people with cystic fibrosis, which is directly proportionate to the country's level of development. On the high end of the range, people with CF can live as long as their mid-50s and 60s. In developing countries, however, the average age of mortality drops to the teenage years.
This is still a vast improvement from years prior to targeted treatments, during which it was rare for someone with cystic fibrosis to live past their 20s.
Matta said current research aims to discover more about CFTR genes and develop new therapeutic technologies to combat the effects of cystic fibrosis.
Kazmerski said the Cystic Fibrosis Foundation supports research related to sexual and reproductive health through the Therapeutics Development Network Sexual Health, Reproduction and Gender Research (SHARING) Working Group. It's important work because the act of sex itself can be difficult for CF sufferers, mostly due to physical discomfort and a lack of libido at times. In addition, male fertility can be affected by CF.
Popular Articles




Related Articles